


FDA 510K (Medical Devices)
 FDA Medical Device Class Ⅰ, Ⅱ, Ⅱ에 따른 인증 및 등록 (510K,PMA)
고객분들의 제품 수출에 있어 사전에 검토해야될 사안들을 꼼꼼이 살피고 면밀하게 분석한 후 의료기기의 용도 및 특성에 따라 Listing, 510K, PMA 등의 절차를 완료해 드립니다.
FDA Medical Device Class Ⅰ, Ⅱ, Ⅱ에 따른 인증 및 등록 (510K,PMA)
고객분들의 제품 수출에 있어 사전에 검토해야될 사안들을 꼼꼼이 살피고 면밀하게 분석한 후 의료기기의 용도 및 특성에 따라 Listing, 510K, PMA 등의 절차를 완료해 드립니다.
[의료기기를 안전성, 유효성에 따라 3등급으로 분류하여 등급에 따라 판매 전 허가 절차를 달리하고 있음]
일반규제요건 : 모든 의료용구에 적용되는 의무사항
공장 및 시설등록(Establishment Registration) : Form 2891을 작성하여 제출
FDA 에서는 미국 내 뿐만 아니라 외국 제조업자에게도 해외공장이나 시설등록을 의무화하고 있음.
해외 제조자들은 미국 내 US Agent를 상주시켜야 하고, US Agent는 제조회사나 제품의 이해 뿐 만 아니라 FDA 법률을 잘 알고 있음으로써, FDA와의 연락책임을 맡아야 함
의료기기등록 (Medical Devices Listing) : Form 2892를 작성하여 제출 의료기기 제조업자는 모든 기기를 각각 등록해야하며, 등록되어 있지 않은 제품은 미국 통관 시 FDA 소속 수입국에 의해 통관 거부되거나 압류됨.
기기등록 역시 미국에 있는 Designated Agent (US Agent)를 통하여 FDA 에 등록되어야 함
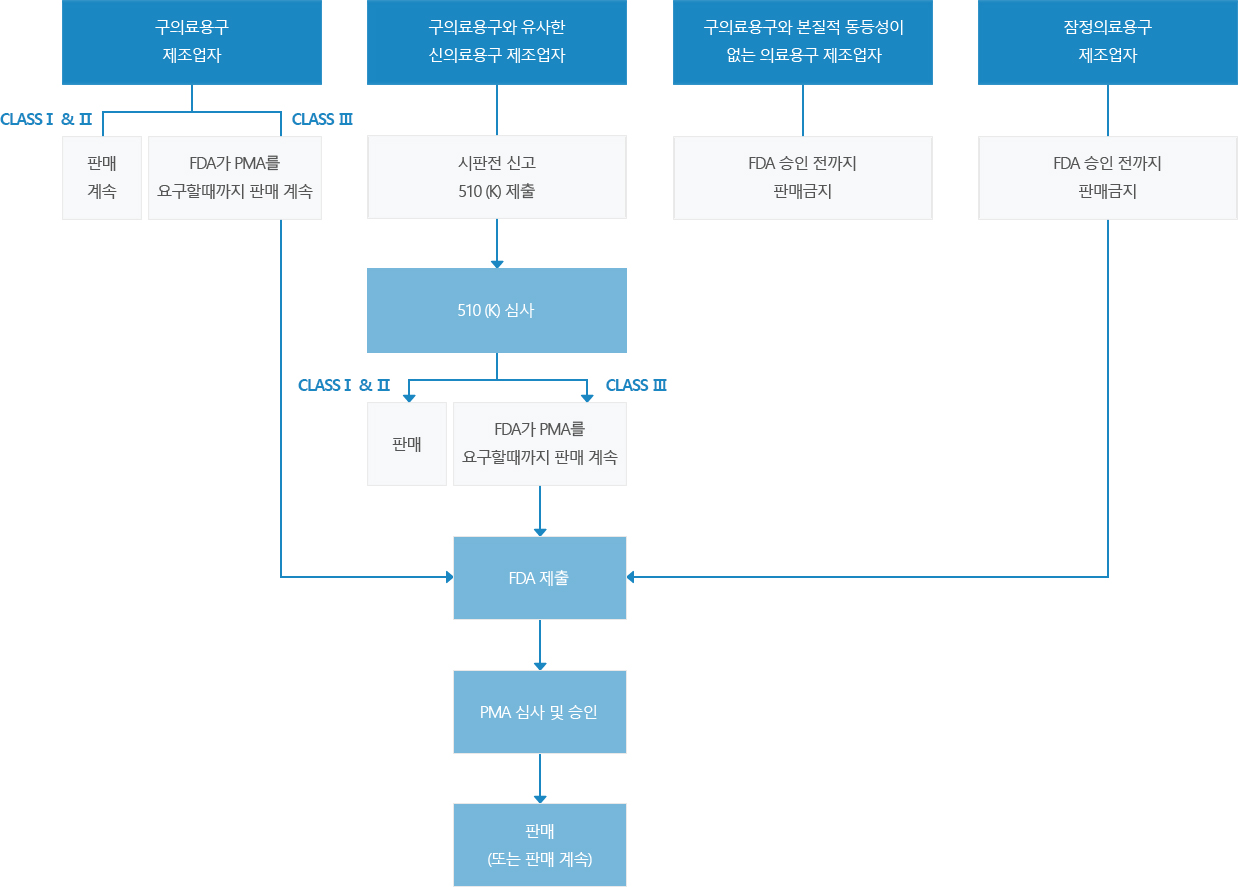
절차
 HOME
FDA Medical Device Class Ⅰ, Ⅱ, Ⅱ에 따른 인증 및 등록 (510K,PMA)
고객분들의 제품 수출에 있어 사전에 검토해야될 사안들을 꼼꼼이 살피고 면밀하게 분석한 후 의료기기의 용도 및 특성에 따라 Listing, 510K, PMA 등의 절차를 완료해 드립니다.
HOME
FDA Medical Device Class Ⅰ, Ⅱ, Ⅱ에 따른 인증 및 등록 (510K,PMA)
고객분들의 제품 수출에 있어 사전에 검토해야될 사안들을 꼼꼼이 살피고 면밀하게 분석한 후 의료기기의 용도 및 특성에 따라 Listing, 510K, PMA 등의 절차를 완료해 드립니다. [의료기기를 안전성, 유효성에 따라 3등급으로 분류하여 등급에 따라 판매 전 허가 절차를 달리하고 있음]
| 1등급 |
인체에 건강과 안전에 심각한 위험을 주지 않는 비교적 단순한 기능의 용구 예) 밴드, 수술용 칼, 수술용 카메라, 썬 텐 부스, 수술용 브러시, 가제, 의료용 솜 등 → 일반규제요건 (General Controls) 적용 시판전 신고 (Premarket Notification 또는 510(k)): 특정제품에 한하기 때문에 미리 제품의 해당여부 확인 |
|---|---|
| 2등급 |
class1 보다 인체의 건강과 안전에 직접적인 영향을 끼칠 수 있는 의료기기들로 일반 통제 검사 이외의 추가요건을 충족 시켜야 함 대부분의 의료기기들이 여기에 해당 예) Bone Cement, 저주파치료기, 레이저수술기, 이식용 클립, Urine Collector, 콘돔, 이식용 척추교정용구, 정형외과용 스테이플, 자동 휠체어, 혈액이나 액체의 주입 펌프, 수술용 멸균된 천 등 일반규제요건 (General Controls) 특별규제요건 (Special Controls) 시판전 신고 (Premarket Notification 또는 510(k)) : 예외의 품목도 있으므로 해당 제품의 확인 필요. |
| 3등급 |
인체의 건강과 안전에 심각한 영향을 끼칠 수 있는 의료용구 들로 classⅠ, Ⅱ의 일반통제는 반드시 거쳐야 함은 물론 판매 전 승인(PMA)도 받아야 함 classⅢ 제품은 일단 통지만으로도 미국 시장에 진출할 수 있는 classⅠ, Ⅱ(II중 일부) 제품과 달리 FDA의 승인을 받은 후에만 미국진출이 가능 예) 이식용 심장밸브, 페이스메이커 건전지, 혈관 팽창용 풍선, 혈관 수술용 레이저, 동맥혈관 접착제, 유방확대용 실리콘, 이식용 뇌촉진기 (implanted cerebella stimulators) 등 일반규제요건 (General Controls) 특별규제요건 (Special Controls) 시판 전 승인 (Premarket Approval, PMA) 임상실험 결과 미국내 제조업자 외에도 미국 내에서 의료용구를 판매하려는 외국 제조업자도 이 기준을 준수하여야며 GMP기준은 의료기기의 제조, 포장, 보관 및 설치에 이용되는 설비와 관리방법에 대하여 규정하고 있음 |
| 의료용구의 제한 | 의료용구의 안전성, 유효성의 합리적 보증이 불가능할 경우 FDA는 그 의료용구의 판매, 유통, 사용을 제한할 수 있음. |
| 등록과 리스트 제출 | 매년 시설등록, 유통하는 의료용구의 리스트 제출 의무 |
해외 제조자들은 미국 내 US Agent를 상주시켜야 하고, US Agent는 제조회사나 제품의 이해 뿐 만 아니라 FDA 법률을 잘 알고 있음으로써, FDA와의 연락책임을 맡아야 함
의료기기등록 (Medical Devices Listing) : Form 2892를 작성하여 제출 의료기기 제조업자는 모든 기기를 각각 등록해야하며, 등록되어 있지 않은 제품은 미국 통관 시 FDA 소속 수입국에 의해 통관 거부되거나 압류됨.
기기등록 역시 미국에 있는 Designated Agent (US Agent)를 통하여 FDA 에 등록되어야 함
| 라벨링 (Labeling) |
라벨링 규정은 제품포장, 설명서나 광고문구등을 포함하며, 제품의 사용방법, 효능, 주의사항 등의 설명이 영문으로 자세히 기재 되어 있어야 함. 허위광고나 과대광고로 오해를 부를 수 있는 문구 또는 제대로 번역 되어 있지 않은 라벨이 있어서는 안 됨 품명, 형명, 제조업자 및 수입자의 성명, 주소기재 |
|---|---|
| 통보 및 수리, 교환과 환불 | 제조업자는 불량 의료용구 발견 즉시 사용자에게 통보하거나, 수리, 교환, 환불할 의무가 있음. |
| 기록과 보고 | 제조업자는 의료용구의 품질을 보증하기 위한 시험검사기록 등을 유지하여야 하며, 필요한 경우 이를 보고할 의무가 있음. |
| 의료용구의 제한 | 의료용구의 안전성, 유효성의 합리적 보증이 불가능할 경우 FDA는 그 의료용구의 판매, 유통, 사용을 제한할 수 있음. |
| 등록과 리스트 제출 | 매년 시설등록, 유통하는 의료용구의 리스트 제출 의무 |